Proteolysis Targeting Chimeras (PROTACs) are heterobifunctional (two-headed) molecules that allow for highly selective targeted protein degradation (TPD). Contrary to traditional small molecule inhibitors (SMIs), PROTACs can degrade disease-causing proteins of interest (POI) that have remained undruggable due to structural challenges, non-enzymatic function, and/or high substrate availability.
PROTACs owe their unique mechanism of action to their structure, composed of a ligand that binds the POI, a flexible polyethylene glycol chemical linker region, and a second ligand that binds to an E3 ubiquitin ligase, hijacking the ubiquitin-proteosome system (UPS). In proximity, both heads are engaged forming a ternary complex (Complex formed between two substrate molecules and an enzyme) where the E3 ligase recruits E2 conjugating enzymes loaded with ubiquitin (Figure 1). Through the action of several E2 conjugating enzymes, poly-ubiquitination of the POI ensues at specific lysine residues/amino acids. Once poly-ubiquitinated, the POI is recognised by the 26S proteosome and subsequently degraded via proteolysis. The PROTAC dissociates in the proteosome and is recycled to degrade other copies of the POI resulting in sub-stochiometric catalysis, in which a single PROTAC can degrade many POI.
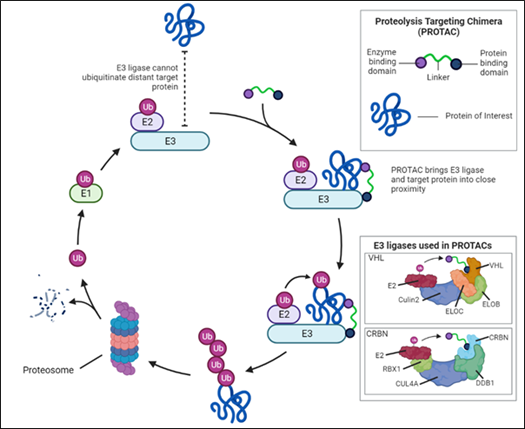
Figure 1.
PROTACs use an event-driven pharmacological mechanism, unlike high-affinity SMIs, which rely on occupancy-driven pharmacology. The therapeutic effectiveness of occupancy-driven approaches hinges on a constant high inhibitory concentration (IC90-95) to block the function of disease-causing proteins (DCPs) which results in off target effects and non-specific binding. However, event-driven pharmacology lowers total cellular levels of DCP, tis allows for greater selectivity and potency even at nM concentrations. Furthermore, their spatial orientation is crucial to form the ternary complex between the POI and the E3 ligase to achieve degradation. Here, PROTAC design is focused on linker optimisation which employs structural information, obtained via cryo-electron microscopy or AI technology like AlphaFold, and target validation via orthogonal analysis through tags fused with the POI. Small changes in the linker region result in dramatic increases in TPD.
The first proof-of-concept of PROTACs was developed by the Crews and Deshaies labs in 2001. PROTAC-1 targeted the protein methionine aminopeptidase-2 (MetAP-2) via an ovalbumin ligand and the SFCβ-TrCP E3 ligase using the endogenous UPS in cellular lysates. Currently, several E3 ligases have been added to the PROTAC toolkit employing a variety of substrate binding domains. The most common examples include: Von Hippel-Lindau (VHL), part of a protein complex formed by elongin B and C, cullin 2 and RBX-1, cereblon (CRBN), part of a Cullin-4-RING E3 ubiquitin ligase, Murine double minute 2 (MDM2) and inhibitor of apoptosis proteins (IAPs) (Figure 1). However, as there are over 600 E3 ligases encoded by the human genome, further development of E3 ligands in PROTACs is required to fully harness/exploit the potential of additional E3 ligases for TPD. PROTACS don’t inherently target specific tissues, as E3 ligases have broad expression profiles. To overcome this hurdle Antibody-PROTAC conjugates such as Ab-PROTAC 3, specific for HER2 positive cells, have the potential to minimise off-target effects.
In total, 18 PROTACs have initiated clinical trials with ARV-110 and ARV-766, both targeting the Androgen Receptor (AR), and in phase II trials for prostate cancer and castration-resistant prostate cancer, respectively. Recently, ARV-471 has entered phase III clinical trials for advanced breast cancer targeting the oestrogen receptor. Despite the possibility of targeting undruggable POIs, PROTAC development has focussed on the AR and Bruton’s Tyrosine Kinase (BTK), both common targets of other drug modalities. In these instances, binding affinity changes due to hotspot mutations, which disproportionately affect traditional SMIs compared to PROTACs which can still fully degrade the POI. Additionally, a low dose regimen and high selectivity conferred by their event-driven pharmacology, reduces off-target effects. Overall, PROTACs can be used for current oncogenic targets.
Although PROTACs are currently in a golden era of funding with a cumulative market capitalisation of 11 billion $, their promise of success faces some challenges. The therapeutic index of PROTACs is highly sensitive to the concentration equilibrium between the POI, E3 ligase and the PROTAC. Accordingly, a hook effect has been observed at supra-optimal ranges where both E3 ligase and the POI are saturated with PROTAC preventing the formation of TDP active ternary complexes. Additionally, as E3 ligases have different expression profiles and functionality in different cell lines, E3 ligand selection must be finely tuned to overcome a poor response and encourage the formation of the ternary complex.
Another, hard to swallow, issue is that their oral bioavailability remains poor due to their large molecular weight, ranging from 0.7 to 1.1 kDa, large polar surface and a high number of hydrogen donors. Lower permeability and solubility of PROTACS augments the extensive cost-of development. To further add to the cost, as complex molecules with intricate pharmacology, early hit PROTACs are inherently hard to predict in terms of their therapeutic utility. To resolve this aspect, enzymology-based approaches may fast-track development and effectively simulate PROTAC pharmacodynamics.
In summary, PROTACs offer a promising therapeutic strategy for TDP of disease-causing proteins. Their unique event-driven pharmacology, potency, and selectivity make them superior to traditional SMIs. However, their development faces challenges including the hook effect, E3 ligand selection, linker optimisation, and poor oral bioavailability. Despite this, the potential of PROTACs is vast and the ongoing clinical trials for prostate cancer demonstrate their ability to degrade oncogenic targets in a real-world setting. Enzymology-based approaches may help in predicting PROTAC pharmacodynamics and streamline their development process. Lastly, targeting previously undruggable targets will require extensive research to characterise the on-target effect of PROTACS.
Comments
If you are a British Pharmacological Society member, please
sign in to post comments.