If there’s one thing that people remember from biology at school, it’s that mitochondria are the powerhouses of the cell. They’re not wrong – in fact, every day the average healthy human produces their bodyweight in the cellular energy currency ATP, and 90 % of this is generated within mitochondria. These credentials make mitochondria undoubtedly essential when it comes to producing energy, however they’re also important for other reasons; these extraordinary organelles contain their own DNA.
Our other genome: why do we care about mitochondrial DNA in the context of precision medicine?
Mitochondrial DNA (mtDNA) is inherited maternally, and populations who share similar mtDNA sequences are referred to as mitochondrial haplogroups. mtDNA is shorter than nuclear DNA (nDNA), containing just 37 genes. Unlike nDNA of which there are two copies per cell in most human cell types, cells can contain thousands of copies of mtDNA, and this number is influenced by the energy demands of the cell. Just like nDNA, variation in mtDNA sequence can influence the way that patients react to drugs. In this age of precision medicine, tailoring treatment to patients’ genetics and lifestyle factors is becoming increasingly common. However, the complex area of mitochondrial pharmacogenetics has been mostly overlooked so far. Arguably the biggest challenge is isolating the effects of mtDNA from nDNA; how can we be sure which genome is responsible?
This is where transmitochondrial cybrids (‘cybrids’) come in. Cybrids are produced by fusing together cells lacking nuclei with cells lacking mtDNA. By incorporating cells with different mtDNA sequences into the same population of mtDNA-devoid cells, we can develop a model where only the mtDNA of a cell changes (and the nDNA remains constant). Taking this approach using the HepG2 liver cell line as the mtDNA-devoid cells has allowed us to generate a model of mtDNA variation which can be used in drug safety screens (Figure 1).
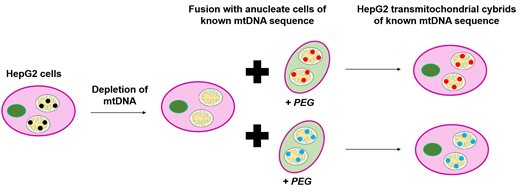
Figure 1. Generation of HepG2 transmitochondrial cybrids. HepG2 cells had their mtDNA depleted by treatment with ethidium bromide (EtBr). Following this, they were combined with cells lacking nuclei (in this case platelets) of known mtDNA sequence in the presence of polyethylene glycol (PEG) to aid fusion. This resulted in the generation of HepG2 transmitochondrial cybrids of varied and known mtDNA sequences all with the same unchanged nDNA.
Good drugs gone bad: potential causes of mitochondrial toxicity
One class of drugs most commonly associated with drug-induced liver injury caused by mitochondrial toxicity are antibiotics, particularly those that target the protein-making machinery (ribosomes) of bacteria. This is because human mitochondrial ribosomes (mitoribosomes) are thought to have evolved from bacterial ribosomes, making them structurally similar. This means that drugs which interfere with bacterial protein synthesis can also disrupt mitochondrial protein synthesis. The essential nature of mitochondria means that this disruption can be very harmful to patients, especially those with mtDNA haplogroups or variants associated with increased susceptibility to toxicity.
One antibiotic which has the potential for off-target inhibition of mitoribosomes is linezolid. Interestingly, the mtDNA variant A2706G has been associated with increased risk of clinical linezolid toxicity, so we investigated to see if we would observe this effect in our model. We used three cell lines: wild-type HepG2 (haplogroup B); HepG2 cybrid H (haplogroup H); and HepG2 cybrid J (haplogroup J) (Table 1).
| Haplogroup |
Variants in region of interest |
| B (HepG2 WT) |
2706G |
| H (HepG2 cybrid) |
|
| J (HepG2 cybrid) |
3010A |
Table 1. HepG2 cell lines used and their variants in the region of interest. The region of interest in this study is the human equivalent of our antibiotics’ bacterial target.
Can transmitochondrial cybrids capture clinical phenomena?
To assess the influence of mtDNA variation on susceptibility to linezolid toxicity, we investigated the effect that 3 days of 100 µM treatment had on the amount of mtDNA and mitochondrial proteins present, as well as the ability of mitochondria to respire and produce ATP. Linezolid exerts its antibacterial effects by preventing initiation of bacterial peptide synthesis (Figure 2). It binds to the 23S ribosomal RNA (rRNA) which normally plays a vital role in the formation of peptide bonds, therefore inhibiting this process. Knowing how linezolid works against bacteria, we hypothesised that there were likely to be decreases in mitoribosome-dependent proteins which would probably induce changes to cellular respiration, but no decreases in the amount of mtDNA as this is upstream of the mitoribosomes (the unsuspecting target of linezolid).
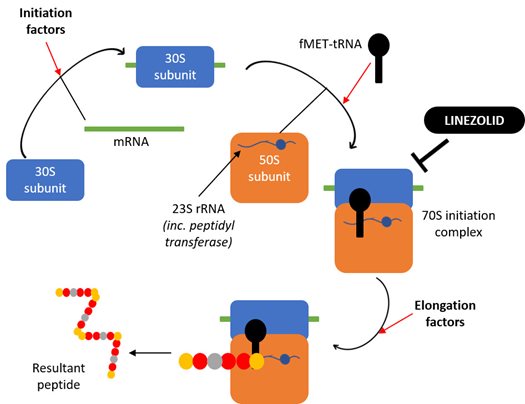
Figure 2. Schematic diagram showing the mechanism of action of the ribosome-targeting antibiotic linezolid against bacterial ribosomes. Linezolid prevents the initiation of bacterial protein synthesis by binding to the 23S rRNA component of the large 50S ribosomal subunit, thereby preventing formation of the initiation complex and inhibiting subsequent peptide synthesis.
To examine the effect of linezolid on mitoribosomes, we compared the expression of two mitochondrial proteins: mitoribosome-dependent protein cyclooxygenase-2 (COXII), and mitoribosome-independent succinate dehydrogenase B (SDHB). As expected, linezolid depleted mitoribosome-dependent proteins in each case. We saw no change in the number of mtDNA copies, which supported our hypothesis that linezolid targets mitochondrial protein expression and not mtDNA replication.
Interestingly, the drastic reduction of mitoribosome-dependent proteins that we found did not lead to a decrease in the rate of ATP production, highlighting the resilience of mitochondria. However, cybrid J had lower respiration rates than HepG2 WT (haplogroup B) and cybrid H which fits with past findings when comparing between haplogroups. We also noticed that linezolid caused significant increases in glycolysis in our cells, which correlates with the fact it can cause abnormally high serum lactate levels in patients (Figure 3). What was most interesting about this increase is that it was most drastic in the cell line containing the A2706G mtDNA variant which is linked with increased risk of linezolid toxicity. This was a really exciting observation, highlighting the ability of this model to capture clinical effects in the lab.
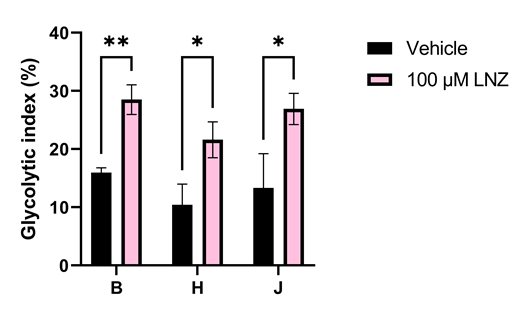
Figure 3. Linezolid increases dependence on glycolysis in all HepG2 cell lines. This increase is most significant in the haplogroup B cell line, which contains the A2706G variant recognised as a risk factor in linezolid toxicity.
Why is this important?
The A2706G mtDNA variant found within HepG2 WT (haplogroup B) cells occurs within the coding region for 16S rRNA, the mitochondrial equivalent of the bacterial 23S rRNA (which is essential for linezolid binding). This variant has been associated with increased risk and severity of linezolid toxicity both clinically and now in our HepG2 cybrid model. As the intended targets of linezolid are bacteria, it is likely that the A2706G mutation increases the similarity of mitoribosomes to bacterial ribosomes. We hope that we have highlighted the importance of considering mtDNA when developing precision medicine strategies.
These findings were used to generate an original research poster, which was awarded first place by the British Pharmacological Society as part of the IUPHAR International Student’s Poster Competition for World Smart Medication Day. Here, we have shown that mitochondrial genome is likely to play a role in patients’ susceptibility to linezolid-induced liver toxicity. The stratification of therapies based on nuclear genes is becoming increasingly common, yet consideration of mtDNA is lacking despite its clinical relevance. To offer one solution to the complex problem of researching links between mtDNA variation and drug safety in a laboratory setting, we provide evidence that HepG2 transmitochondrial cybrids are a useful model of mtDNA variation in drug safety studies. These results form part of a larger project led by Dr Amy Chadwick at the University of Liverpool.
Comments
If you are a British Pharmacological Society member, please
sign in to post comments.